
Analysis in relationships of overlapping genes in systemic sclerosis-associated interstitial lung disease, idiopathic pulmonary fibrosis and pulmonary fibrosis model induced by transforming growth factor-β
-
摘要:目的
利用生物信息学技术分析系统性硬化症相关间质性肺病(SSc-ILD)、特发性肺纤维化(IPF)与转化生长因子-β(TGF-β)诱导的肺纤维化模型重叠基因的相关性。
方法通过Gene Expression Omnibus(GEO)数据库获取SSc-ILD、IPF与TGF-β诱导的肺纤维化模型相关基因芯片数据GSE76808、GSE135099。采用R软件对数据进行分析,按照adj.P<0.05和|logFC|>1进行筛选,将筛选得到的基因进行基因交互分析;利用R软件对重叠基因进行基因本体(GO)功能注释、京都基因与基因组百科全书(KEGG)通路富集及可视化;基于String数据库对重叠基因进行蛋白质互作(PPI)分析;应用在线分析工具TISIDB获得hub基因并分析其功能。
结果与健康对照组(NC组)相比,SSc-ILD、IPF、TGF-β诱导的肺纤维化模型中存在差异性表达的基因。在SSc-ILD、IPF、TGF-β诱导的肺纤维化模型均下调的34个重叠基因中存在PPI并获得hub基因ELAVL1。GO富集分析结果表明,ELAVL1基因不仅参与RNA剪接、加工、翻译等多种转录后修饰过程,还在腺苷酸活化蛋白激酶(AMPK)信号通路中存在富集。
结论在SSc-ILD和IPF两种疾病中,AMPK信号可能存在异常,导致TGF-β产生增多,而TGF-β可能通过调控ELAVL1蛋白(HuR)由胞核向胞浆转位,进而参与肺纤维化的调节,但具体机制仍有待进一步验证。
-
关键词:
- 转化生长因子-β /
- 系统性硬化症相关间质性肺病 /
- 特发性肺纤维化 /
- 生物信息学分析
Abstract:ObjectiveTo analyze the relationships of overlapping genes in systemic sclerosis-associated interstitial lung disease (SSc-ILD), idiopathic pulmonary fibrosis (IPF) and pulmonary fibrosis model induced by transforming growth factor-β (TGF-β) by using bioinformatics technology.
MethodsRelevant gene chip data GSE76808 and GSE135099 of SSc-ILD, IPF and pulmonary fibrosis model induced by TGF-β were obtained from Gene Expression Omnibus (GEO) database. R software was used to analyze the data, and according to the screening conditions of adj. P<0.05 and |logFC|>1, the screened genes were analyzed by gene interaction; Gene Ontology (GO) function annotation, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment and visualization for overlapping genes were performed by using R software; protein-protein interaction (PPI) analysis of overlapping genes was performed based on string database; the hub gene was obtained by analysis tool TISIDB and its function was analyzed.
ResultsCompared with the normal control group (NC group), there were differentially expressed genes in SSc-ILD, IPF and pulmonary fibrosis model induced by TGF-β. There was PPI existed in 34 overlapping genes down-regulated by SSc-ILD, IPF and pulmonary fibrosis model induced by TGF-β, and hub gene ELAVL1 was obtained. GO enrichment analysis results showed that ELAVL1 gene was not only involved in many post-transcriptional modification processes such as RNA splicing, processing and translation, but also enriched in adenylate activated protein kinase (AMPK) signal pathway.
ConclusionIn SSc-ILD and IPF, the AMPK signal may be abnormal, resulting in increasing of TGF-β, and TGF-β may be involved in the regulation of pulmonary fibrosis by regulating the translocation of ELAVL1 protein (HuR) from nucleus to cytoplasm, but the specific mechanism still needs to be further verified.
-
转化生长因子-β(TGF-β)是转化生长因子超家族中的一员,在调节细胞分化、增殖、迁移、免疫反应和细胞外基质(ECM)沉积中发挥至关重要的作用[1]。TGF-β通常以前体的形式合成,需要进一步激活以触发其下游的经典或非经典途径[2-3]。研究[4-6]表明, TGF-β可通过激活经典的Smads通路来促进纤维化相关分子转录,包括α-平滑肌肌动蛋白(α-SMA)、Ⅰ型胶原和基质金属蛋白酶组织抑制剂(TIMP), 其可诱导成纤维细胞活化和ECM沉积,导致组织器官纤维化。TGF-β介导的非经典的Smads信号通路也可以上调促纤维化因子的表达,导致ECM过度产生,参与到纤维化过程中[7]。系统性硬化症(SSc)是一种局限性或弥漫性皮肤及内脏结缔组织纤维化、硬化及萎缩的结缔组织疾病,该病较罕见,女性多见。30%的SSc患者可并发间质性肺病(ILD)。ILD是SSc患者最常见的死亡原因, 10年病死率高达40%[8-11]。SSc的发病机制较为复杂,其中TGF-β及相关细胞因子启动的细胞信号转导可促进SSc纤维化的形成[12]。TGF-β通路可直接作用于成纤维细胞,促进2型先天淋巴样细胞向促纤维化表型转化,导致白细胞介素-10 (IL-10)产生减少,胶原合成增加,促进SSc纤维化[13]。
TGF-β生成增加不仅能诱导成纤维细胞向肌成纤维细胞分化,还可导致细胞内钙调节失调,上调纤维化标志物α-SMA、纤维连接蛋白和波形蛋白的表达,加重SSc肺纤维化[14]。特发性肺纤维化(IPF)是一种病因不明的慢性、进行性、纤维化性间质性肺病,诊断IPF后,患者的中位生存期为2~4年[15]。目前研究[16]认为肺纤维化是肺泡上皮反复微损伤的结果。肺泡上皮细胞在修复过程中,释放过多的TGF-β等促纤维化因子[17]。促纤维化介质激活并促进驻留的成纤维细胞分化为肌成纤维细胞,产生过量的ECM[18]。成纤维细胞/肌成纤维细胞的聚集和ECM的沉积,破坏了正常的肺结构,阻碍了气体交换,进而加重IPF[19]。虽然上述两种疾病的具体发病机制尚不清楚,但TGF-β信号的异常均可诱发或促进系统性硬化症间质性肺病(SSc-ILD)和IPF发展。本研究分析SSc-ILD、IPF、TGF-β诱导的肺纤维化模型重叠基因的相关性,探讨TGF-β介导的信号通路是否可以作为SSc-ILD和IPF共同的致病途径,现报告如下。
1. 材料与方法
1.1 研究材料
本研究利用Gene Expression Omnibus (GEO)数据库(https://www.ncbi.nlm.nih.gov/geo/)[20]获取GSE76808、GSE135099基因芯片数据,研究类型均为Expression profiling by array, 2个数据源分别在GPL570和GPL13534平台上检测
1.2 研究方法
1.2.1 数据处理
通过R软件“tidyverse”包将GSE76808、GSE135099原始数据中的“ID-REF”转换为“Gene Symbol”。使用R软件的“limma”包筛选出差异性表达的基因,筛选标准为adj.P value<0.05和|logFC|>1。利用R软件将差异基因进行处理并绘制火山图。
1.2.2 获取重叠基因
对差异基因交互制作韦恩图,利用R软件对重叠基因进行基因本体(GO)功能注释、京都基因与基因组百科全书(KEGG)通路富集及可视化。
1.2.3 构建蛋白质互作(PPI)网络并筛选分析hub基因
从String数据库[21]搜索重叠基因间的相互作用关系,将PPI关系导入Cytoscape构建PPI网络,根据Degree值将重叠基因排序,利用“GO”插件筛选出hub基因。通过在线网站TISIDB(http://cis.hku.hk/TISIDB/)[22]对hub基因功能和通路富集进行分析
2. 结果
2.1 SSc-ILD、IPF、TGF-β诱导的肺纤维化模型中差异性表达的基因
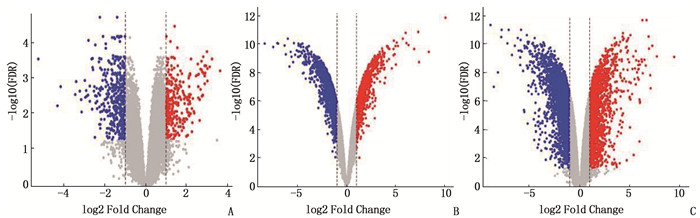
本研究对GEO数据库收录的GSE76808、GSE135099数据进行差异分析发现,与健康对照组(NC组)相比, SSc-ILD中有279个基因表达上调,362个基因表达下调; 与NC组相比, IPF中存在820个高表达的基因, 1 350个低表达的基因; 与NC组相比, TGF-β诱导的肺纤维化模型中存在1 562个上调的基因, 2 416个下调的基因。见图 1。
![]() 图 1 SSc-ILD、IPF、TGF-β诱导的肺纤维化模型中差异性表达的基因A: 与NC组相比, SSc-ILD中差异性表达的基因; B: 与NC组相比, IPF中差异性表达的基因; C: 与NC组相比, TGF-β诱导的肺纤维化模型中差异性表达的基因。
图 1 SSc-ILD、IPF、TGF-β诱导的肺纤维化模型中差异性表达的基因A: 与NC组相比, SSc-ILD中差异性表达的基因; B: 与NC组相比, IPF中差异性表达的基因; C: 与NC组相比, TGF-β诱导的肺纤维化模型中差异性表达的基因。2.2 SSc-ILD差异性表达的基因与TGF-β诱导的肺纤维化模型差异性表达的基因中的重叠基因
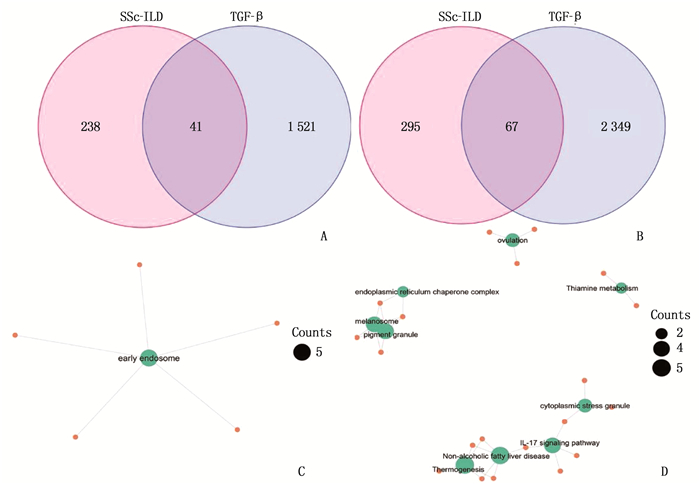
将上述筛选出的SSc-ILD中279个高表达基因与TGF-β诱导的肺纤维化模型中1 562个高表达基因进行交互分析,得到41个上调的重叠基因; SSc-ILD中362个低表达基因与TGF-β诱导的肺纤维化模型中的2 416个低表达基因交互结果显示,有67个下调的重叠基因。对上述重叠基因进行GO分析发现,重叠的上调基因可能参与了早期内体的形成; 重叠的下调基因可能在白细胞介素-17(IL-17)信号通路、非酒精性脂肪肝病等方面具有重要的调节功能。见图 2。
![]() 图 2 SSc-ILD差异性表达的基因与TGF-β诱导的肺纤维化模型差异性表达的基因中的重叠基因A、B: 分别为SSc-ILD与TGF-β诱导的肺纤维化模型中高表达、低表达基因的重叠基因韦恩图; C、D: 分别为SSc-ILD与TGF-β诱导的肺纤维化模型中高表达、低表达的重叠基因GO分析结果。
图 2 SSc-ILD差异性表达的基因与TGF-β诱导的肺纤维化模型差异性表达的基因中的重叠基因A、B: 分别为SSc-ILD与TGF-β诱导的肺纤维化模型中高表达、低表达基因的重叠基因韦恩图; C、D: 分别为SSc-ILD与TGF-β诱导的肺纤维化模型中高表达、低表达的重叠基因GO分析结果。2.3 IPF差异性表达的基因与TGF-β诱导的肺纤维化模型差异性表达的基因中的重叠基因
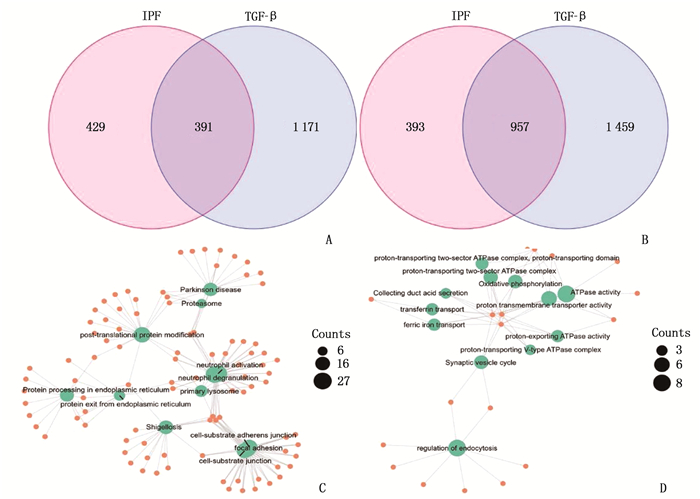
对筛选出的IPF中820个上调的基因与TGF-β诱导的肺纤维化模型中的1 562个上调基因进行交互,获得391个高表达的重叠基因; IPF中下调的1 350个基因与TGF-β诱导的肺纤维化模型中的2 416个下调基因进行交互,获得957个低表达的重叠基因。对上述重叠基因进行GO分析发现,重叠的上调基因主要富集在中性粒细胞激活、中性粒细胞脱颗粒、蛋白翻译后修饰、蛋白在内质网内的加工与运输等方面; 重叠的下调基因可能在氧化磷酸化、ATP酶活性等方面发挥关键的调节作用。见图 3。
![]() 图 3 IPF差异性表达的基因与TGF-β诱导的肺纤维化模型差异性表达的基因中的重叠基因A、B: 分别为IPF与TGF-β诱导的肺纤维化模型差异表达基因中的重叠基因韦恩图; C、D: 分别为IPF与TGF-β诱导的肺纤维化模型中上调和下调的重叠基因GO分析结果。
图 3 IPF差异性表达的基因与TGF-β诱导的肺纤维化模型差异性表达的基因中的重叠基因A、B: 分别为IPF与TGF-β诱导的肺纤维化模型差异表达基因中的重叠基因韦恩图; C、D: 分别为IPF与TGF-β诱导的肺纤维化模型中上调和下调的重叠基因GO分析结果。2.4 SSc-ILD、IPF、TGF-β诱导的肺纤维化模型之间的重叠基因
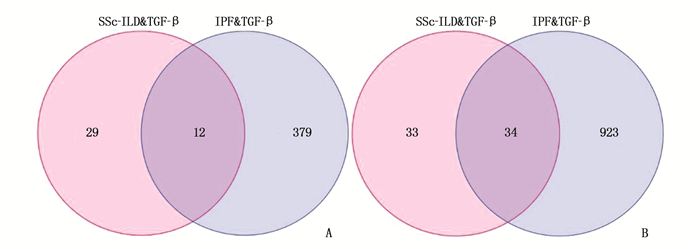
分别将SSc-ILD&TGF-β重叠基因与IPF&TGF-β重叠基因进行交互,发现在上调基因中存在12个重叠基因,在下调基因中存在34个重叠基因。见图 4。
![]() 图 4 SSc-ILD、IPF、TGF-β诱导的肺纤维化模型之间的重叠基因韦恩图A: SSc-ILD、IPF、TGF-β诱导的肺纤维化模型之间共同上调基因的韦恩图; B: SSc-ILD、IPF、TGF-β诱导的肺纤维化模型之间共同下调基因的韦恩图。
图 4 SSc-ILD、IPF、TGF-β诱导的肺纤维化模型之间的重叠基因韦恩图A: SSc-ILD、IPF、TGF-β诱导的肺纤维化模型之间共同上调基因的韦恩图; B: SSc-ILD、IPF、TGF-β诱导的肺纤维化模型之间共同下调基因的韦恩图。2.5 重叠基因的PPI分析
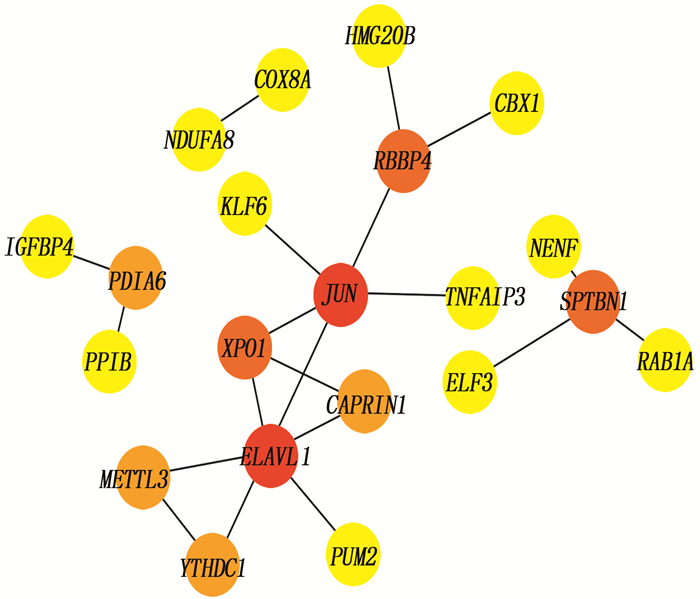
对SSc-ILD、IPF、TGF-β诱导的肺纤维化模型之间的重叠基因进行PPI网络分析发现,上调12个基因在蛋白水平没有出现相互作用,下调的34个基因中存在PPI并得到hub基因 ELAVL1 。见图 5、表 1。
表 1 PPI网络分析中前21个基因排序 名称 得分 排序 名称 得分 1 ELAVL1 6 10 RAB1A 1 2 JUN 5 10 COX8A 1 3 RBBP4 3 10 NDUFA8 1 3 SPTBN1 3 10 PUM2 1 3 XPO1 3 10 ELF3 1 6 METTL3 2 10 HMG20B 1 6 YTHDC1 2 10 IGFBP4 1 6 PDIA6 2 10 TNFAIP3 1 6 CAPRIN1 2 10 KLF6 1 10 PPIB 1 10 NENF 1 10 CBX1 1 2.6 GO和KEGG对hub基因功能和通路的分析
GO分析结果显示, ELAVL1 基因参与RNA剪接、加工、翻译等多种转录后修饰过程见表 2、表 3; KEGG分析结果显示, ELAVL1 基因仅在腺苷酸活化蛋白激酶(AMPK)信号通路中存在富集。
表 2 GO分析 ELAVL1 基因GO号 生物过程 GO号 生物过程 GO号 生物过程 0000375 通过酯交换反应进行RNA剪接 0031047 RNA基因沉默 0048255 mRNA稳定 0000377 通过酯交换反应与凸起的腺苷作为亲核试剂进行RNA剪接 0034248 细胞内酰胺代谢过程的调节 0060147 转录后基因沉默的调控 0000398 通过剪接体进行mRNA剪接 0034249 细胞内酰胺代谢过程的负调控 0060149 转录后基因沉默的负调控 0006397 mRNA加工 0034250 细胞内酰胺代谢过程的正向调控 0060964 miRNA对基因沉默的调控 0006417 调节翻译 0035194 RNA转录后基因沉默 0060965 miRNA对基因沉默的负调控 0008380 RNA剪接 0035195 miRNA的基因沉默 0060966 RNA对基因沉默的调控 0010608 基因表达的转录后调控 0040029 基因表达调控、表观遗传 0060967 RNA对基因沉默的负调控 0016441 转录后基因沉默 0043487 RNA稳定性的调节 0060968 基因沉默的调控 0016458 基因沉默 0043488 mRNA稳定性的调节 0060969 基因沉默的负调控 0017148 翻译的负面调节 0043489 RNA稳定 0070935 3′-UTR介导的mRNA稳定性 0019827 维持干细胞群 0045727 正向调节翻译 0098727 维持细胞数量 表 3 GO分析 ELAVL1 基因GO号 分子功能 0003725 结合双链RNA 0003729 结合mRNA 0003730 结合mRNA 3′-UTR 0017091 结合富含AU元素 0035925 结合mRNA 3′-UTR富含AU元素区域 3. 讨论
肺纤维化是多种间质性肺病的终末期,会引起不可逆性肺损伤,其特征是正常肺泡结构被破坏, ECM过度沉积导致患者肺部气体交换受阻,呼吸功能不全,甚至死亡。SSc-ILD和IPF患者在人口学特征和致病因素上有所不同,但这两种疾病均可导致肺实质纤维化,最终进展为呼吸衰竭,其中持续存在的异常TGF-β信号在肺纤维化中起着关键作用[23]。SSc-ILD中,先天免疫系统和获得性免疫系统的过度激活可导致血管内皮细胞和肺泡上皮细胞损伤。同时, TGF-β信号的异常激活可诱导内皮细胞和上皮细胞间充质转化,以及成纤维细胞向肌成纤维细胞的转化,导致ECM过度积聚,促进SSc-ILD[24]。IPF的形成起源于肺泡上皮的慢性损伤和(或)老化,导致异常的伤口愈合反应以及持续产生促炎和促纤维化因子,这最终导致血管周围和间质内间充质细胞的活化和分化。TGF-β信号升高是IPF的重要标志,其可促进成纤维细胞向肌成纤维细胞转化,上调胶原蛋白相关基因的表达,导致ECM产生增多[25]。
家族性肺纤维化的存在和罕见的遗传性疾病中肺纤维化的发生促使研究人员关注到宿主遗传背景在影响SSC-ILD和IPF患者病程和病情发展中的重要作用[26]。2019年由10 000名欧洲患者组成的全基因组关联研究(GWAS)确定了23个与SSc相关的独立基因座,使得已知的SSc风险基因座总数增加到28个,并且这些SSc风险位点相关的大多数基因产物与炎症和自身免疫有关[27]。此外,与普通人群相比, SSc患者和家庭聚集性强的患者的一级亲属罹患SSc的风险更高,估计风险约为1.6%, 表明SSc具有基因遗传倾向[28]。在IPF中,端粒相关基因(TERT、TERC、PARN、 RTEL1 、 DKC1 、 TINF2 )、表面活性物质编码基因(SFTPC、 SFTPA1 、 SFTPA2 )的罕见变异和20个独立基因座的单核苷酸多态性(SNPs)在散发性IPF疾病风险中增高,占25%~30%, 并且在IPF的发病机制中,与这些位点相关的基因在对端粒完整性、细胞黏附、纤维化和宿主防御途径方面扮演着重要的角色[29-30]。基于TGF-β在纤维化过程、SSc-ILD和IPF中的作用,以及SSC-ILD和IPF发生的基因遗传背景,本研究利用GEO数据库获取SSc-ILD、TGF-β和IPF相关基因数据,通过生物信息学技术对上述数据分析发现, SSc-ILD、IPF分别与TGF-β诱导的纤维化模型存在相同的基因。SSc-ILD与TGF-β诱导的纤维化模型共同上调的基因参与早期内体的形成,而共同下调的基因在调节IL-17信号通路中发挥重要作用。IPF与TGF-β诱导的纤维化模型共同的上调基因参与了中性粒细胞激活、中性粒细胞脱颗粒、蛋白翻译后修饰、加工与运输等的调节; 共同的下调基因可能在氧化磷酸化、ATP酶活性等过程中发挥关键作用。TGF-β信号可能通过调控基因的上述功能分别参与SSC-ILD和IPF的发病过程。
本研究将SSc-ILD&TGF-β重叠基因与IPF&TGF-β重叠基因进行交互,发现存在12个重叠高表达基因; 在下调基因中存在34个重叠基因。上述重叠基因的PPI分析发现,在重叠的12个高表达基因中不存在蛋白互作,但在34个低表达基因中存在蛋白互作,并获得hub基因 ELAVL1 。 ELAVL1 基因可以编码胚胎致死性异常视觉(ELAV)家族中的人抗原R(HuR), 即ELAVL1蛋白,其是一种RNA结合蛋白(RBP)。HuR广泛表达于多种组织中,通过转录后机制参与调控细胞内多种分子的表达[31]。在静息细胞中, HuR主要定位于细胞核,当受到紫外线辐射、营养耗竭或免疫激活等应激条件的刺激时, HuR结合并将靶mRNA运送到细胞质,起到调节靶mRNA的稳定性和(或)翻译的作用[32]。研究[33]发现, HuR能够调节促炎细胞因子(肿瘤坏死因子-α、白细胞介素-6、转化生长因子-β和干扰素-γ)、促炎介质INOS和趋化因子 MCP-1 mRNA的表达,这些因素大多参与了肝纤维化的发病过程。TGF-β通过影响HuR对mRNA稳定性和翻译率的调节在肝纤维化发展中的基因表达调控方面起着重要作用。本研究GO功能分析同样发现, ELAVL1 基因参与调节RNA剪接、加工、翻译等多种转录后修饰; 同时, ELAVL1 能够与双链DNA、mRNA结合,发挥至关重要的分子功能,表明 ELAVL1 基因可能通过编码ELAVL1蛋白(HuR)参与SSC-ILD和IPF多种纤维化分子mRNA转录后调控。
在IPF中,人肺成纤维细胞经TGF-β处理后, HuR由胞核向胞浆转位明显增加。在肺纤维化小鼠模型中,肺组织细胞的胞浆HuR显著高于对照组。HuR可能通过促进肌成纤维细胞的分化以及增加易感个体ECM的产生参与肺纤维化[34]。本研究TGF-β诱导的纤维化模型、IPF和SSc-ILD中均出现差异性表达的 ELAVL1 基因,表明TGF-β可能通过诱导IPF与SSc-ILD肺成纤维细胞中的HuR从胞核向胞浆移动,调节纤维化相关因子mRNA的表达,导致ECM产生异常,参与两种疾病的发展。AMPK是多条代谢途径的中枢调节因子,作为TGF-β/Smads通路的上游信号分子,对成纤维细胞的增殖、分化和ECM的产生的调节是必不可少的[35]。血管紧张素Ⅱ可诱导心肌纤维化并伴有TGF-β过度表达、Smad2/3高磷酸化和Smad4表达上调。黄芩苷可通过激活AMPK, 抑制TGF-β/Smads通路,从而抑制成纤维细胞增殖和ECM积聚,缓解心肌纤维化[36]。本研究KEGG通路分析发现, ELAVL1 基因富集在AMPK信号通路中。一项探究HuR是否参与气道平滑肌增殖的研究[37]指出, HuR是唯一一个在真核细胞中普遍表达的蛋白; HuR的表达或活性可能受到多种上游刺激的影响,如炎症介质、生长因子、活性氧蔟(ROS)和缺氧。另外,激活AMPK可有效抑制血小板衍生生长因子(PDGF)诱导的HuR胞浆易位,揭示了ELAVL1蛋白与AMPK信号通路之间的潜在联系。因此,作者推测在SSc-ILD和IPF中, AMPK信号异常可能导致TGF-β产生增多,过度产生的TGF-β可能通过诱导HuR由胞核向胞浆转位,进而参与肺纤维化的调节。但确切的调节机制尚不清楚,需要进一步开展大样本的研究,对本研究中的关键结论进行验证,为阐明SSc-ILD和IPF共同的发病机制提供科学依据。
-
![]()
图 1 SSc-ILD、IPF、TGF-β诱导的肺纤维化模型中差异性表达的基因
A: 与NC组相比, SSc-ILD中差异性表达的基因; B: 与NC组相比, IPF中差异性表达的基因; C: 与NC组相比, TGF-β诱导的肺纤维化模型中差异性表达的基因。
![]()
图 2 SSc-ILD差异性表达的基因与TGF-β诱导的肺纤维化模型差异性表达的基因中的重叠基因
A、B: 分别为SSc-ILD与TGF-β诱导的肺纤维化模型中高表达、低表达基因的重叠基因韦恩图; C、D: 分别为SSc-ILD与TGF-β诱导的肺纤维化模型中高表达、低表达的重叠基因GO分析结果。
![]()
图 3 IPF差异性表达的基因与TGF-β诱导的肺纤维化模型差异性表达的基因中的重叠基因
A、B: 分别为IPF与TGF-β诱导的肺纤维化模型差异表达基因中的重叠基因韦恩图; C、D: 分别为IPF与TGF-β诱导的肺纤维化模型中上调和下调的重叠基因GO分析结果。
![]()
图 4 SSc-ILD、IPF、TGF-β诱导的肺纤维化模型之间的重叠基因韦恩图
A: SSc-ILD、IPF、TGF-β诱导的肺纤维化模型之间共同上调基因的韦恩图; B: SSc-ILD、IPF、TGF-β诱导的肺纤维化模型之间共同下调基因的韦恩图。
表 1 PPI网络分析中前21个基因
排序 名称 得分 排序 名称 得分 1 ELAVL1 6 10 RAB1A 1 2 JUN 5 10 COX8A 1 3 RBBP4 3 10 NDUFA8 1 3 SPTBN1 3 10 PUM2 1 3 XPO1 3 10 ELF3 1 6 METTL3 2 10 HMG20B 1 6 YTHDC1 2 10 IGFBP4 1 6 PDIA6 2 10 TNFAIP3 1 6 CAPRIN1 2 10 KLF6 1 10 PPIB 1 10 NENF 1 10 CBX1 1  下载: 导出CSV
下载: 导出CSV
表 2 GO分析 ELAVL1 基因
GO号 生物过程 GO号 生物过程 GO号 生物过程 0000375 通过酯交换反应进行RNA剪接 0031047 RNA基因沉默 0048255 mRNA稳定 0000377 通过酯交换反应与凸起的腺苷作为亲核试剂进行RNA剪接 0034248 细胞内酰胺代谢过程的调节 0060147 转录后基因沉默的调控 0000398 通过剪接体进行mRNA剪接 0034249 细胞内酰胺代谢过程的负调控 0060149 转录后基因沉默的负调控 0006397 mRNA加工 0034250 细胞内酰胺代谢过程的正向调控 0060964 miRNA对基因沉默的调控 0006417 调节翻译 0035194 RNA转录后基因沉默 0060965 miRNA对基因沉默的负调控 0008380 RNA剪接 0035195 miRNA的基因沉默 0060966 RNA对基因沉默的调控 0010608 基因表达的转录后调控 0040029 基因表达调控、表观遗传 0060967 RNA对基因沉默的负调控 0016441 转录后基因沉默 0043487 RNA稳定性的调节 0060968 基因沉默的调控 0016458 基因沉默 0043488 mRNA稳定性的调节 0060969 基因沉默的负调控 0017148 翻译的负面调节 0043489 RNA稳定 0070935 3′-UTR介导的mRNA稳定性 0019827 维持干细胞群 0045727 正向调节翻译 0098727 维持细胞数量
下载: 导出CSV
表 3 GO分析 ELAVL1 基因
GO号 分子功能 0003725 结合双链RNA 0003729 结合mRNA 0003730 结合mRNA 3′-UTR 0017091 结合富含AU元素 0035925 结合mRNA 3′-UTR富含AU元素区域
下载: 导出CSV
-
[1] ZHANG X L, YUN J S, HAN D, et al. TGF-β pathway in salivary gland fibrosis[J]. Int J Mol Sci, 2020, 21(23): E9138. doi: 10.3390/ijms21239138
[2] MORIKAWA M, DERYNCK R, MIYAZONO K. TGF-β and the TGF-β family: context-dependent roles in cell and tissue physiology[J]. Cold Spring Harb Perspect Biol, 2016, 8(5): a021873. doi: 10.1101/cshperspect.a021873
[3] HATA A, CHEN Y G. TGF-β signaling from receptors to smads[J]. Cold Spring Harb Perspect Biol, 2016, 8(9): a022061. doi: 10.1101/cshperspect.a022061
[4] LAMOUILLE S, XU J, DERYNCK R. Molecular mechanisms of epithelial-mesenchymal transition[J]. Nat Rev Mol Cell Biol, 2014, 15(3): 178-196. doi: 10.1038/nrm3758
[5] TRIPATHI V, SIXT K M, GAO S J, et al. Direct regulation of alternative splicing by SMAD3 through PCBP1 is essential to the tumor-promoting role of TGF-Β[J]. Mol Cell, 2016, 64(3): 549-564. doi: 10.1016/j.molcel.2016.09.013
[6] BUDI E H, DUAN D N, DERYNCK R. Transforming growth factor-β receptors and smads: regulatory complexity and functional versatility[J]. Trends Cell Biol, 2017, 27(9): 658-672. doi: 10.1016/j.tcb.2017.04.005
[7] MENG X M, NIKOLIC-PATERSON D J, LAN H Y. TGF-β: the master regulator of fibrosis[J]. Nat Rev Nephrol, 2016, 12(6): 325-338. doi: 10.1038/nrneph.2016.48
[8] ELHAI M, MEUNE C, BOUBAYA M, et al. Mapping and predicting mortality from systemic sclerosis[J]. Ann Rheum Dis, 2017, 76(11): 1897-1905. doi: 10.1136/annrheumdis-2017-211448
[9] KHEDOE P, MARGES E, HIEMSTRA P, et al. Interstitial lung disease in patients with systemic sclerosis: toward personalized-medicine-based prediction and drug screening models of systemic sclerosis-related interstitial lung disease (SSc-ILD)[J]. Front Immunol, 2020, 11: 1990. doi: 10.3389/fimmu.2020.01990
[10] PERELAS A, SILVER R M, ARROSSI A V, et al. Systemic sclerosis-associated interstitial lung disease[J]. Lancet Respir Med, 2020, 8(3): 304-320. doi: 10.1016/S2213-2600(19)30480-1
[11] DENTON C P, KHANNA D. Systemic sclerosis[J]. Lancet, 2017, 390(10103): 1685-1699. doi: 10.1016/S0140-6736(17)30933-9
[12] CABRAL-MARQUES O, RIEMEKASTEN G. Vascular hypothesis revisited: role of stimulating antibodies against angiotensin and endothelin receptors in the pathogenesis of systemic sclerosis[J]. Autoimmun Rev, 2016, 15(7): 690-694. doi: 10.1016/j.autrev.2016.03.005
[13] LAURENT P, ALLARD B, MANICKI P, et al. TGFβ promotes low IL10-producing ILC2 with profibrotic ability involved in skin fibrosis in systemic sclerosis[J]. Ann Rheum Dis, 2021, 80(12): 1594-1603. doi: 10.1136/annrheumdis-2020-219748
[14] HSU W L, HSIEH Y C, YU H S, et al. 2-Aminoethyl diphenylborinate inhibits bleomycin-induced skin and pulmonary fibrosis via interrupting intracellular Ca2+ regulation[J]. J Dermatol Sci, 2021, 103(2): 101-108. doi: 10.1016/j.jdermsci.2021.07.005
[15] GEORGE P M, PATTERSON C M, REED A K, et al. Lung transplantation for idiopathic pulmonary fibrosis[J]. Lancet Respir Med, 2019, 7(3): 271-282. doi: 10.1016/S2213-2600(18)30502-2
[16] RICHELDI L, COLLARD H R, JONES M G. Idiopathic pulmonary fibrosis[J]. Lancet, 2017, 389(10082): 1941-1952. doi: 10.1016/S0140-6736(17)30866-8
[17] AL-TAMARI H M, DABRAL S, SCHMALL A, et al. FoxO3 an important player in fibrogenesis and therapeutic target for idiopathic pulmonary fibrosis[J]. EMBO Mol Med, 2018, 10(2): 276-293. doi: 10.15252/emmm.201606261
[18] HOSSEINZADEH A, JAVAD-MOOSAVI S A, REITER R J, et al. Idiopathic pulmonary fibrosis (IPF) signaling pathways and protective roles of melatonin[J]. Life Sci, 2018, 201: 17-29. doi: 10.1016/j.lfs.2018.03.032
[19] KOLB M, BONELLA F, WOLLIN L. Therapeutic targets in idiopathic pulmonary fibrosis[J]. Respir Med, 2017, 131: 49-57. doi: 10.1016/j.rmed.2017.07.062
[20] EDGAR R, DOMRACHEV M, LASH A E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository[J]. Nucleic Acids Res, 2002, 30(1): 207-210. doi: 10.1093/nar/30.1.207
[21] SZKLARCZYK D, MORRIS J H, COOK H, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible[J]. Nucleic Acids Res, 2017, 45(D1): D362-D368. doi: 10.1093/nar/gkw937
[22] RU B B, WONG C N, TONG Y, et al. TISIDB: an integrated repository portal for tumor-immune system interactions[J]. Bioinformatics, 2019, 35(20): 4200-4202. doi: 10.1093/bioinformatics/btz210
[23] STOCK C J W, MICHAELOUDES C, LEONI P, et al. Bromodomain and extraterminal (BET) protein inhibition restores redox balance and inhibits myofibroblast activation[J]. Biomed Res Int, 2019, 2019: 1484736.
[24] VALENZI E, TABIB T, PAPAZOGLOU A, et al. Disparate interferon signaling and shared aberrant basaloid cells in single-cell profiling of idiopathic pulmonary fibrosis and systemic sclerosis-associated interstitial lung disease[J]. Front Immunol, 2021, 12: 595811. doi: 10.3389/fimmu.2021.595811
[25] DECARIS M L, SCHAUB J R, CHEN C, et al. Dual inhibition of αvβ6 and αvβ1 reduces fibrogenesis in lung tissue explants from patients with IPF[J]. Respir Res, 2021, 22(1): 265. doi: 10.1186/s12931-021-01863-0
[26] DISTLER J H W, GYÖRFI A H, RAMANUJAM M, et al. Shared and distinct mechanisms of fibrosis[J]. Nat Rev Rheumatol, 2019, 15(12): 705-730. doi: 10.1038/s41584-019-0322-7
[27] ANGIOLILLI C, MARUT W, VAN DER KROEF M, et al. New insights into the genetics and epigenetics of systemic sclerosis[J]. Nat Rev Rheumatol, 2018, 14(11): 657-673. doi: 10.1038/s41584-018-0099-0
[28] TRUCHETET M E, BREMBILLA N C, CHIZZOLINI C. Current concepts on the pathogenesis of systemic sclerosis[J]. Clin Rev Allergy Immunol, 2021: 1-13.
[29] ALLEN R J, GUILLEN-GUIO B, OLDHAM J M, et al. Genome-wide association study of susceptibility to idiopathic pulmonary fibrosis[J]. Am J Respir Crit Care Med, 2020, 201(5): 564-574. doi: 10.1164/rccm.201905-1017OC
[30] LORENZO-SALAZAR J M, MA S F, JOU J, et al. Novel idiopathic pulmonary fibrosis susceptibility variants revealed by deep sequencing[J]. ERJ Open Res, 2019, 5(2): 00071-02019.
[31] WANG J, GUO Y, CHU H L, et al. Multiple functions of the RNA-binding protein HuR in cancer progression, treatment responses and prognosis[J]. Int J Mol Sci, 2013, 14(5): 10015-10041. doi: 10.3390/ijms140510015
[32] ZHANG F, CAI Z L, LV H D, et al. Multiple functions of HuR in urinary tumors[J]. J Cancer Res Clin Oncol, 2019, 145(1): 11-18. doi: 10.1007/s00432-018-2778-2
[33] WOODHOO A, IRUARRIZAGA-LEJARRETA M, BERAZA N, et al. Human antigen R contributes to hepatic stellate cell activation and liver fibrosis[J]. Hepatology, 2012, 56(5): 1870-1882. doi: 10.1002/hep.25828
[34] AL-HABEEB F, ALOUFI N, TRABOULSI H, et al. Human antigen R promotes lung fibroblast differentiation to myofibroblasts and increases extracellular matrix production[J]. J Cell Physiol, 2021, 236(10): 6836-6851. doi: 10.1002/jcp.30380
[35] GAO L, WANG L Y, LIU Z Q, et al. TNAP inhibition attenuates cardiac fibrosis induced by myocardial infarction through deactivating TGF-β1/Smads and activating P53 signaling pathways[J]. Cell Death Dis, 2020, 11(1): 44. doi: 10.1038/s41419-020-2243-4
[36] XIAO Y C, YE J T, ZHOU Y, et al. Baicalin inhibits pressure overload-induced cardiac fibrosis through regulating AMPK/TGF-β/Smads signaling pathway[J]. Arch Biochem Biophys, 2018, 640: 37-46. doi: 10.1016/j.abb.2018.01.006
[37] ZHANG P J, CAO M F, LIU Y, et al. PDGF-induced airway smooth muscle proliferation is associated with Human antigen R activation and could be weakened by AMPK activation[J]. Mol Biol Rep, 2012, 39(5): 5819-5829. doi: 10.1007/s11033-011-1392-z
-
期刊类型引用(3)
1. 宋丹,许蕊. 特发性肺纤维化患者血清FSTL1、Syndecan-1与肺功能和预后的关系. 检验医学与临床. 2024(04): 450-454 .  百度学术
百度学术
2. 张静,毛英. 血清转化生长因子β1、白细胞介素-6、Toll样受体-4、核转录因子κB联合评估非小细胞肺癌放射性肺炎病情严重程度的价值. 实用临床医药杂志. 2024(14): 12-17 . 本站查看
3. 丁达庆,孔晓梅. 药物治疗进展性纤维化性间质性肺疾病的新进展与思考. 现代医学与健康研究电子杂志. 2023(21): 138-141 . 百度学术
其他类型引用(1)
计量
- 文章访问数: 328
- HTML全文浏览量: 204
- PDF下载量: 47
- 被引次数: 4
 苏公网安备 32100302010246号
苏公网安备 32100302010246号