
Advances for ferroptosis in treating myocardial ischemia reperfusion injury
-
摘要:
铁死亡是一种铁依赖性的脂质过氧化介导细胞膜损伤致细胞死亡的新方式, 其受多种细胞代谢途径的调控, 包括铁代谢、脂质代谢、氧化还原系统等, 许多器官的损伤和退行性病变均与其相关, 在治疗缺血性疾病和脂质过氧化相关的退行性变疾病中有巨大潜力。心肌缺血再灌注损伤(MIRI)是急性心肌梗死患者进行血运重建治疗后最常见的死亡原因, 近年的研究表明铁死亡与MIRI密切相关, 通过氧化应激、铁代谢、脂质代谢、内质网应激、炎症反应等影响MIRI, 干预再灌注过程中的铁死亡可以有效改善心功能, 减少梗死面积。本文就铁死亡在MIRI中的具体作用及相关研究进展予以综述。
Abstract:Ferroptosis, a new form of programmed cell death marked by iron-dependent phospholipid peroxidation, is regulated by complex cellular metabolic pathways, including iron metabolism, lipid metabolism, and oxidation-reduction system, is associated with many organ injuries and degeneration, and has great potential in the treatment of ischemic diseases and lipid peroxide-related degenerative diseases. Myocardial ischemia reperfusion injury (MIRI) is the most common cause of death in patients with acute myocardial infarction after revascularization therapy. Recent studies have shown that ferroptosis is intimately related to the pathological process of MIRI. Ferroptosis is associated with MIRI through oxidative stress, iron metabolism, lipid metabolism, endoplasmic reticulum stress and inflammatory response. Intervention of ferroptosis during reperfusion can effectively improve cardiac function and reduce myocardial infarct size. In this paper, the research progress was explored between ferroptosis and MIRI, and the specific role of ferroptosis in MIRI was discussed.
-
铁死亡作为一种新的细胞死亡形式在各种器官缺血再灌注损伤、神经退行性疾病、恶性肿瘤中扮演重要角色,其中对于心肌缺血再灌注损伤(MIRI)的相关研究尤其突出。MIRI是指冠状动脉部分或完全急性阻塞后,在一定时间内重新获得再通时,缺血心肌虽然得以恢复正常灌注,但其组织损伤反而呈进行性加重的病理过程[1]。多种化合物可以通过作用于谷胱甘肽过氧化物酶4(GPX4)、铁代谢、脂质代谢等铁死亡核心调控途径来改善MIRI[2]。本文就铁死亡在MIRI中的具体机制及研究进展进行综述,以期为治疗MIRI提供参考。
1. 铁死亡机制
1.1 以GPX4为核心的Xc-GSH-GPX4途径
Xc系统是一种由溶质载体家族7成员11(SLC7A11)和溶质载体家族3成员2(SLC3A2) 2个亚基组成,分布于磷脂双分子层的氨基酸反转运蛋白。胱氨酸和谷氨酸(Glu)通过Xc系统以1∶ 1的比例在细胞内外进行交换,胱氨酸被吸收入细胞中还原为半胱氨酸(Cys), 参与谷胱甘肽(GSH)的合成。谷胱甘肽过氧化物酶4(GPX4)催化脂质氢过氧化物(L-OOH)与GSH的巯基结合,将GSH氧化为氧化型谷胱甘肽(GSSG),并将L-OOH还原为无毒的脂质醇(L-OH), 从而阻断ROS链反应,抑制铁死亡。这就是经典的Xc-GSH-GPX4途径,抑制其中任何一个步骤都会增加脂质过氧化物的积累导致铁死亡。核因子E2相关因子2(NRF2)直接作用于Xc系统和GPX4,激活抗氧化系统,抑制NRF2的表达会极大促进铁死亡的发生[3]。RSL3作为经典的铁死亡诱导剂,可以直接作用于GPX4并抑制其活性,从而降低细胞的抗氧化能力导致铁死亡[4]。P53可以通过下调SLC7A11的表达来抑制胱氨酸的摄取,从而影响GPX4的活性,导致细胞抗氧化能力降低、ROS积累,并最终发生铁死亡[5]。外源性的Cu2+能选择性地与GPX4蛋白上的半胱氨酸残基C107和C148相结合,促进GPX4的寡聚化,随后依赖Tax1结合蛋白1 (TAX1BP1)促进GPX4的自噬降解,提高细胞对铁死亡的敏感性[6]。
1.2 不依赖GPX4的新途径
GPX4相关途径是铁死亡的经典核心调控机制,此外亦有不依赖GPX4的新途径,包括FSP1-CoQ10-NADPH通路、DHODH-CoQ10通路、GCH1-BH4通路。铁死亡抑制蛋白1 (FSP1)具有辅酶Q10(CoQ10)氧化还原酶的活性,还原形式的辅酶Q10(CoQ10-H2)是磷脂和脂蛋白中的一种良好的抗氧化剂,CoQ10-H2清除脂质过氧化物后被氧化为CoQ10, FSP1通过消耗NADPH将CoQ10还原为CoQ10-H2, 如此, FSP1-CoQ10-NADPH通过不断减少脂质过氧化物的积累发挥抗铁死亡作用[7]。二氢乳清酸脱氢酶(DHODH)是一种线粒体内膜蛋白,亦可以通过将CoQ10还原成CoQ10-H2在线粒体内膜中发挥抗氧化能力抵抗铁死亡[8]。四氢生物蝶呤(BH4)是一种有效的自由基捕获抗氧化剂(RTA), 由三磷酸鸟苷(GTP)在环GTP水解酶1(GCH1)作用下生成,BH4与二氢生物蝶呤(BH2)配对形成氧化还原循环,在二氢叶酸还原酶(DHFR)作用下不断再生,可以减少内源性氧化自由基的积累,从而保护细胞膜以抑制铁死亡。在此过程中抑制GCH1的表达或促进DHFR的降解都能显著增加细胞对铁死亡的敏感程度[9]。
1.3 铁代谢
铁离子过度积累是铁死亡的重要特征,亚铁离子(Fe2+)经肠道吸收或经红细胞降解形成,随后被氧化成铁离子(Fe3+), Fe3+与转铁蛋白(TF)结合,被转铁蛋白受体1(TFR-1)摄取,在细胞内被金属还原酶(STEAP3)还原为Fe2+参与多种生理过程,此外多余的Fe2+会储存在不稳定铁池(LIP)中。LIP中的Fe2+参加芬顿反应并生成大量的羟自由基,大量的羟自由基能与细胞膜、质膜中的多不饱和脂肪酸(PUFA)反应生成大量的脂质过氧化物,造成细胞铁死亡,这是细胞铁死亡的另一条经典途径。热休克蛋白β-1(HSPB1)可以抑制TFR-1的表达水平,从而减少细胞内LIP含量;去铁胺(DFO)是一种铁螯合剂,能够通过细胞内吞作用堆积在细胞溶酶体内,并螯合细胞内游离铁,阻止铁向氧化物传递电子,以此减少活性氧的生成[10]。HSPB1和DFO都是从控制铁摄入方面抑制铁死亡,而铁外排和铁储存方面对铁死亡也有很大的影响。铁泵蛋白1(FPN1)是目前哺乳动物细胞中已知的唯一一类铁外排蛋白, FPN1的过表达同样可以减少LIP含量从而发挥抑制铁死亡的作用[11]。核受体共激活因子4 (NCOA4)能够直接识别并结合铁蛋白重链1(FTH1), 然后将铁蛋白输送到自噬体进行溶酶体降解,丝氨酸/苏氨酸蛋白激酶(Ser/Thr PK)能促进NCOA4-FTH1之间的相互作用,加剧铁蛋白自噬介导的铁死亡[12], 而化合物9a(铁卟啉抑制剂)通过破坏NCOA4-FTH1的相互作用发挥抗铁死亡作用[13]。
1.4 脂质代谢
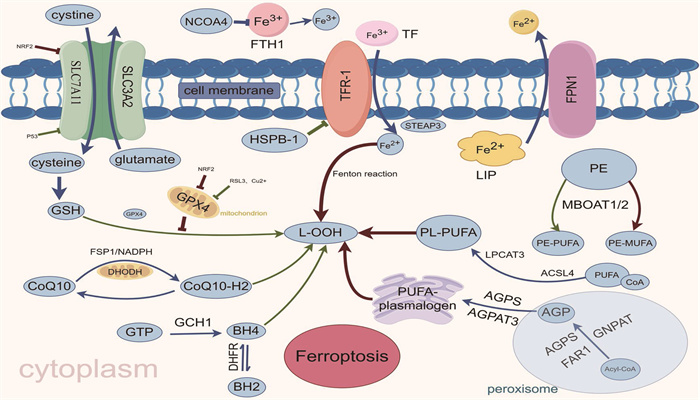
在各种类型的细胞脂质中,磷脂(PL)、鞘脂和胆固醇共同构成细胞膜的主要成分。铁死亡主要是由PL过氧化引起,其中PL过氧化的首要底物是含有多不饱和脂肪酸的磷脂酰乙醇胺(PE-PUFA), 而膜结合O-酰基转移酶1/2(MBOAT1/2)可以重塑细胞磷脂谱,选择性地将单不饱和脂肪酸(MUFA)转移到溶血磷脂酰乙醇胺(lyso-PE)中,从而增加细胞中的含单不饱和脂肪酸的磷脂酰乙醇胺(PE-MUFA), 减少细胞中含有多不饱和脂肪酸的磷脂酰乙醇胺(PE-PUFA)来抑制铁死亡[14]。此外,有证据[15]表明鞘脂和胆固醇也参与铁死亡过程,抑制鞘脂合成可显著减少Erastin或谷氨酸诱导的海马神经元细胞铁死亡,抑制胆固醇合成可抑制恶性淋巴瘤细胞铁死亡。长链脂酰辅酶A合成酶4(ACSL4)是脂质过氧化所需要的一种脂质代谢酶, ACSL4将长链PUFA与CoA连接形成长链酰基CoA, 进而由溶血卵磷脂酰基转移酶3(LPCAT3)将长链酰基CoA插入PL中。ACSL4和LPCAT3都参与铁死亡脂质代谢的信号传导,敲除细胞中的 ACSL4 和 LPCAT3 基因,可以显著抑制细胞铁死亡,并且通过药理学靶点的筛选发现,有部分药物[如罗格列酮(ROSI)、吡格列酮(PIO)和曲格列酮(TRO)等] 可以通过作用于ACSL4和LPCAT3来抑制铁死亡,有效改善神经变性、肝坏死,以及脑、肾、肝和心脏等组织的缺血再灌注损伤[16-17]。在铁死亡中除了ACSL4/LPCAT3介导的脂质代谢途径,还有另一条依赖于过氧化物酶体的脂质代谢途径,即酰基辅酶A (Acyl-CoA)在烷基甘油磷酸合成酶(AGPS)、脂肪酰基辅酶A还原酶1(FAR1)和甘油磷酸O-酰基转移酶(GNPAT)作用下合成前体1-O-烷基甘油-3-磷酸(AGP), 然后通过1-酰基甘油-3-磷酸-O-酰基转移酶3(AGPAT3)和缩醛磷脂乙醇胺去饱和酶1(PEDS1)运到内质网形成多不饱和脂肪酸缩醛磷脂(PUFA-plasmalogen),促进细胞脂质过氧化[18]。见图 1。
![]() 图 1 铁死亡主要调控机制GSH: 谷胱甘肽; GSSG: 氧化型谷胱甘肽; GPX4: 谷胱甘肽过氧化物酶4; Glutamate: 谷氨酸;
图 1 铁死亡主要调控机制GSH: 谷胱甘肽; GSSG: 氧化型谷胱甘肽; GPX4: 谷胱甘肽过氧化物酶4; Glutamate: 谷氨酸;
SLC7A11: 溶质载体家族7成员11; SLC3A2: 溶质载体家族3成员2; Cysteine: 半胱氨酸; Cystine: 胱氨酸;
NRF2: 核因子E2相关因子2; L-OOH: 脂质过氧化物; L-OH: 脂质醇; CoQ10-H2: 还原形式的辅酶Q10;
FSP1: 铁死亡抑制蛋白1; DHODH: 二氢乳清酸脱氢酶; GCH1: 三磷酸鸟苷环化水解酶1; BH4: 四氢生物蝶呤;
BH2: 二氢生物蝶呤; GTP: 三磷酸鸟苷; DHFR: 二氢叶酸还原酶; MBOAT1/2: 膜结合O-酰基转移酶1/2;
MUFA: 单不饱和脂肪酸; PUFA: 多不饱和脂肪酸; ACSL4: 长链脂酰辅酶A合成酶4;
LPCAT3: 溶血卵磷脂酰基转移酶3; LIP: 不稳定铁池; TF: 转铁蛋白; TFR-1: 转铁蛋白受体1;
核NCOA4: 受体共激活因子4; FTH1: 铁蛋白重链1; HSPB1: 热休克蛋白β-; Acyl-CoA: 酰基辅酶A;
AGPS: 烷基甘油磷酸合成酶; FAR1: 脂肪酰基辅酶A还原酶1; GNPAT: 甘油磷酸O-酰基转移酶;
AGP: 前体1-O-烷基甘油-3-磷酸; AGPAT3: 1-酰基甘油-3-磷酸-O-酰基转移酶3;
PEDS1: 缩醛磷脂乙醇胺去饱和酶1; PUFA-plasmalogen: 多不饱和脂肪酸缩醛磷脂。2. MIRI机制
MIRI分为再灌注性心律失常(RA)、心肌顿抑、冠状动脉微血管阻塞(MVO)、致死性心肌再灌注损伤4种类型[19], 主要与氧化应激、炎症反应、钙离子超载、线粒体渗透性转换孔(MPTP)通道开放、再灌注时PH值快速恢复等机制相关。
2.1 氧化应激
机体在心肌再灌注的最初几分钟内产生大量ROS, 导致氧化系统和抗氧化系统之间失衡并产生多种毒副作用。首先,过量的ROS引发脂质过氧化,改变细胞的结构和功能完整性; 其次,ROS刺激核因子κB (NF-κB)活化进一步触发心肌炎症。针对此的许多抗氧化药物(维生素C/E、多酚类药物、抗坏血酸、N-乙酰半胱氨酸、去铁胺等)在临床应用的效果褒贬不一,需要更多的研究来推动抗氧化疗法的进步[20]。
2.2 炎症反应
MIRI引起的炎症反应是无菌性炎症,在此过程中损伤的心肌释放趋化因子吸引中性粒细胞进入梗死区,中性粒细胞激活分子通路产生多种炎性因子,这是一把双刃剑,一方面能够促进细胞碎片的清除和伤口愈合,但另一方面损伤相关分子模式(DAMP)介导的信号传导会加剧炎症状态,导致组织进一步损伤[21]。同时,活化的中性粒细胞在缺血组织中产生NADPH氧化酶,引起呼吸暴发和髓过氧化物酶的释放并进一步造成组织损伤。
2.3 钙离子超载
当心肌缺血时血氧供应中断,能量供应转为无氧糖酵解,导致心肌细胞内H+积累,此时过度激活质膜上的Na+/H+交换器, Na+的过量内流反过来激活Na+/Ca2+交换器导致Ca2+内流。再灌注时,胞外H+的消耗进一步提高了质子膜上的质子梯度且内质网钙泵会进一步促进Ca2+的摄取,从而增加胞质内Ca2+的含量。一方面,钙超载后会引起MPTP开放,导致线粒体膜电位异常、凋亡因子释放等引起心肌细胞死亡; 另一方面,钙超载可激活钙蛋白酶分解肌原纤维,造成心肌进一步损伤[22]。
2.4 线粒体渗透性转换孔(MPTP)通道的开放
MPTP是位于线粒体内外膜的非选择性通道, MPTP通道的打开会引起线粒体的一系列改变,如通透性的转变、内电位的丢失、呼吸链的中断、基质肿胀和线粒体膜破裂等。MPTP在心肌缺血期间保持关闭,再灌注时开放,以响应线粒体内钙离子过载、氧化应激、ATP消耗以及pH值的快速恢复[19]。在再灌注开始时,给予MPTP抑制剂(免疫抑制剂环孢素A), 可有效减少40%~50%的梗死面积[23]。
2.5 再灌注时pH值的快速恢复
急性心肌缺血时,细胞内pH值迅速降至7.0以下,再灌注时血氧恢复,堆积的乳酸被迅速冲洗, Na+/H+交换体及Na+-HCO3-转运体激活, pH值迅速恢复。在再灌注的最初几分钟,这种pH值的迅速变化导致MPTP开放和心肌细胞内Ca2+超载,致使心肌过度挛缩并最终导致心肌细胞死亡。用酸性缓冲液灌注缺血心脏、药物抑制Na+/H+交换器或通过减缓心肌再灌注过程(缺血后适应)等都可以有效减少梗死面积[24]。
3. 铁死亡在心肌缺血再灌注损伤中的作用
过氧化磷脂酰乙醇胺(PEox)是铁死亡的预测生物标志物,SPARVERO L J等[25]应用气体团簇离子束次生离子质谱(GCIB-SIMS)在H9C2细胞中直接检测到PEox, 这为心肌细胞铁死亡的发生提供了直接证据。动物实验中发现在MIRI的再灌注期发生铁死亡,并随再灌注时间的延长加重[26], 在临床中无论是慢性冠心病(CAD)还是ST段抬高型心肌梗死(STEMI)患者中,再灌注前对患者加用DFO可显著减轻心肌氧化应激损伤并改善患者心功能[27]。因此,不论是动物试验或是临床验证,均有充足的证据证明铁死亡在MIRI的重要作用。
3.1 抗氧化系统
铁死亡的核心分子机制是氧化系统和抗氧化系统之间的失衡,过度的氧化应激和氧化还原系统的破坏导致产生过多的脂质过氧化物,进一步导致线粒体膜密度增加、线粒体嵴断裂和线粒体质膜的破坏,导致细胞铁死亡。Xc-GSH-GPX4轴是铁死亡中最重要的抗氧化系统。GPX-4是线粒体上游重要的抗氧化酶,调节铁死亡和氧化应激, MIRI大鼠心脏中Fe2+和丙二醛(MDA)水平随着再灌注时间的延长而逐渐升高,而GPX-4水平随之下降[26], 铁死亡抑制剂(liproxstatin-1)可以上调GPX-4表达水平抑制铁死亡,减轻再灌注损伤。P53是体内重要的抑癌基因,其在MIRI期间高表达, P53作用于SLC7A11来抑制Xc-GSH-GPX4轴诱导铁死亡,而泛素特异性蛋白酶22(USP22)通过作用于NAD依赖性蛋白去乙酰化酶sirtuin-1 (SIRT1)抑制P53的转录活性,抑制MIRI中的铁死亡[5]。NRF2是体内重要的抗氧化元件,激活NRF2相关通路可以减轻MIRI, 依托咪酯(etomidate)通过诱导MIRI大鼠心肌细胞内NRF2的核易位及其下游靶基因血红素加氧合酶1(HO-1)的过表达,激活NRF2/HO-1通路上调GSH活性、促进GPX4表达来抑制再灌注诱导的铁死亡减轻心肌损伤[28]。但另有研究[29]表明, HO-1可能通过增加细胞内铁含量,促进Erastin诱导的肿瘤细胞脂质过氧化促进铁死亡。
3.2 铁过载
无论在心肌缺血或再灌注期间,铁蛋白的降解都强于铁蛋白合成,随着缺血时间的延长,冠脉中铁离子水平较缺血前大幅升高,尤其是再灌注后细胞内环境的酸性和高还原性进一步促进铁蛋白释放Fe3+, 从而增加Fe2+含量促进芬顿反应,生成更多的过氧化物造成铁死亡,加剧心肌再灌注损伤,因此减轻心脏铁负荷抑制心肌细胞铁死亡可以显著改善MIRI。心肌缺血前的预处理可显著提高铁蛋白含量,降低Fe3+水平,减轻铁过载介导的心肌再灌注损伤,影响铁蛋白自噬会影响MIRI的铁代谢, NCOA4的表达量在缺血再灌注处理的心肌组织中上调[30], DNA甲基转移酶1 (DNMT-1)通过抑制NCOA4介导的铁蛋白自噬来减少心肌细胞内铁离子含量[31]。除铁蛋白外,其他与铁代谢相关的蛋白质也参与了MIRI的过程。在缺氧/再复氧(H/R)心肌细胞中外泌体(exosome)作用的研究中, Exo抑制转铁蛋白受体(TFR)的表达,减少心肌细胞中铁的摄取和积累,减轻H/R诱导的心肌细胞损伤[32]。在大鼠MIRI模型中,通过泛素特异性蛋白酶7 (USP7)/P53/TFR1通路, I/R损伤会上调USP7的表达来减少P53泛素化降解,从而增加TFR1表达以促进心肌细胞游离铁的摄取,造成心肌细胞铁死亡[33]。Hepcidin是一种细胞内铁调节蛋白,可降解FPN1致细胞内铁过载,而在心脏缺血缺氧时Hepcidin蛋白的表达会上调,这可能与缺氧诱导因子(HIF)的表达相关[34]。
3.3 脂质过氧化
MIRI的发生伴随着脂质代谢异常。ACSL4和LPCAT3是铁死亡中脂质代谢通路的关键酶,其表达量在I/R心肌组织显著升高,促进ACSL4的泛素化降解会显著改善I/R心肌细胞的铁死亡[35]。除此之外,脂氧合酶(ALox15)催化花生四烯酸(AA)过氧化,促进铁死亡,在心肌组织会因I/R损伤过表达, 15-氢过氧二十碳四烯酸(15-HpETE)是ALox15促进AA过氧化中重要的中间产物,通过促进受体γ辅助激活因子1-α(Pgc1α)的泛素化降解,导致线粒体脂质过氧化水平升高、功能障碍、形态异常,最终导致心肌细胞铁死亡,而特异性地抑制Alox15, 可以显著提高Pgc1α蛋白水平,抑制心肌细胞铁死亡,改善小鼠再灌注损伤后的心功能[36]。
3.4 内质网应激
内质网是蛋白质合成、加工、修饰的关键位点,在维持细胞稳态方面发挥着重要作用,在缺血缺氧时内质网中未折叠蛋白质的积累或错误折叠,会激活未折叠蛋白反应(UPR), 将应激信号传入细胞核,此过程称为内质网应激(ERS)[19]。青蒿琥酯(ART)是一种铁死亡诱导剂,作用于活化转录因子4(ATF4)介导的URP, ART通过ATF4-CHOP(C/EBP同源蛋白)-PUMA(P53细胞凋亡上调调节剂)-P53途径使P53过表达促进脂质过氧化物的产生,其可以诱导肝癌细胞过氧化致肝癌细胞铁死亡[37]。在MIRI期间,ERS反应增加,ERS中的未折叠蛋白会诱导心肌损伤,最近发现在加用Ferrostatin-1后可显著减少ERS和心肌细胞损伤[38]。
3.5 炎症反应
梗死心肌会释放DAMP激活nod样受体蛋白3炎症小体(NLRP3), NLRP3活化半胱氨酸天冬氨酸酶-1(caspase-1)诱导促炎因子IL-1β和IL-18的成熟、释放,从而激活无菌免疫反应[21]。人冠状动脉标本免疫组化染色结果显示,NLRP3的表达量与前列腺素内过氧化物合酶2(PTGS2)和ACSL4呈正相关,而PTGS2和ACSL4是铁死亡过程中的重要成分[39]。Xc-GSH-GPX4轴是铁死亡中最重要的抗氧化系统, GSH、GPX4可以抑制NLRP3的激活,减少炎症介质的释放,在Ferrostatin-1处理过的细胞中, NLRP3的活性也会显著下降[40]。最近的一项猪心脏骤停复苏后心脏损伤的研究同样发现,抑制NLRP3的激活可以改善心脏损伤,同时降低细胞铁死亡水平[41]。此外DAMP还能通过Toll样受体4(TLR4)激活NF-κB, 导致一系列炎性细胞因子的释放,而目前许多研究都表明铁死亡与NF-κB通路密切相关。富马酸二甲酯(DMF)作为NRF2的激活剂,通过NRF2/ARE/NF-κB信号通路,促进关键的铁死亡因子血红素加氧酶1(HO-1)、NADPH醌氧化还原酶1(NQO1)和GPX4的表达,增加细胞对铁死亡的抵抗力[42]。
4. 结论
综上所述,铁死亡作为一种细胞内铁依赖性的细胞死亡形式,通过多种方式参与MIRI, 包括抗氧化系统、铁代谢、脂质代谢、内质网应激、炎症反应等。因此,对铁死亡作用机制及其相应干预措施的深入探索是当前研究的重点,如调控铁死亡的关键因子NRF2, 作为经典的抗氧化元件对铁死亡的调控表现出两面性,对此仍需进一步探索。此外,铁死亡与坏死、凋亡等细胞死亡方式的作用机制相互交织,如何选择性地调控铁死亡是未来研究的重点。
-
![]()
图 1 铁死亡主要调控机制
GSH: 谷胱甘肽; GSSG: 氧化型谷胱甘肽; GPX4: 谷胱甘肽过氧化物酶4; Glutamate: 谷氨酸;
SLC7A11: 溶质载体家族7成员11; SLC3A2: 溶质载体家族3成员2; Cysteine: 半胱氨酸; Cystine: 胱氨酸;
NRF2: 核因子E2相关因子2; L-OOH: 脂质过氧化物; L-OH: 脂质醇; CoQ10-H2: 还原形式的辅酶Q10;
FSP1: 铁死亡抑制蛋白1; DHODH: 二氢乳清酸脱氢酶; GCH1: 三磷酸鸟苷环化水解酶1; BH4: 四氢生物蝶呤;
BH2: 二氢生物蝶呤; GTP: 三磷酸鸟苷; DHFR: 二氢叶酸还原酶; MBOAT1/2: 膜结合O-酰基转移酶1/2;
MUFA: 单不饱和脂肪酸; PUFA: 多不饱和脂肪酸; ACSL4: 长链脂酰辅酶A合成酶4;
LPCAT3: 溶血卵磷脂酰基转移酶3; LIP: 不稳定铁池; TF: 转铁蛋白; TFR-1: 转铁蛋白受体1;
核NCOA4: 受体共激活因子4; FTH1: 铁蛋白重链1; HSPB1: 热休克蛋白β-; Acyl-CoA: 酰基辅酶A;
AGPS: 烷基甘油磷酸合成酶; FAR1: 脂肪酰基辅酶A还原酶1; GNPAT: 甘油磷酸O-酰基转移酶;
AGP: 前体1-O-烷基甘油-3-磷酸; AGPAT3: 1-酰基甘油-3-磷酸-O-酰基转移酶3;
PEDS1: 缩醛磷脂乙醇胺去饱和酶1; PUFA-plasmalogen: 多不饱和脂肪酸缩醛磷脂。 -
[1] PREM P N, SIVAKUMAR B, BOOVARAHAN S R, et al. Recent advances in potential of Fisetin in the management of myocardial ischemia-reperfusion injury-a systematic review[J]. Phytomedicine, 2022, 101: 154123. doi: 10.1016/j.phymed.2022.154123
[2] ZHAO W K, ZHOU Y, XU T T, et al. Ferroptosis: opportunities and challenges in myocardial ischemia-reperfusion injury[J]. Oxid Med Cell Longev, 2021, 2021: 9929687.
[3] YANG X, HUANG T Y, CHEN Y H, et al. Deoxynivalenol induces testicular ferroptosis by regulating the Nrf2/System Xc-/GPX4 axis[J]. Food Chem Toxicol, 2023, 175: 113730. doi: 10.1016/j.fct.2023.113730
[4] CUI Y, ZHANG Z L, ZHOU X, et al. Microglia and macrophage exhibit attenuated inflammatory response and ferroptosis resistance after RSL3 stimulation via increasing Nrf2 expression[J]. J Neuroinflammation, 2021, 18(1): 249. doi: 10.1186/s12974-021-02231-x
[5] MA S X, SUN L Y, WU W H, et al. USP22 protects against myocardial ischemia-reperfusion injury via the SIRT1-p53/SLC7A11-dependent inhibition of ferroptosis-induced cardiomyocyte death[J]. Front Physiol, 2020, 11: 551318. doi: 10.3389/fphys.2020.551318
[6] XUE Q, YAN D, CHEN X, et al. Copper-dependent autophagic degradation of GPX4 drives ferroptosis[J]. Autophagy, 2023, 19(7): 1982-1996. doi: 10.1080/15548627.2023.2165323
[7] LI W T, LIANG L, LIU S Y, et al. FSP1: a key regulator of ferroptosis[J]. Trends Mol Med, 2023, 29(9): 753-764. doi: 10.1016/j.molmed.2023.05.013
[8] MAO C, LIU X G, ZHANG Y L, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer[J]. Nature, 2021, 593(7860): 586-590. doi: 10.1038/s41586-021-03539-7
[9] HU Q, WEI W H, WU D Q, et al. Blockade of GCH1/BH4 axis activates ferritinophagy to mitigate the resistance of colorectal cancer to erastin-induced ferroptosis[J]. Front Cell Dev Biol, 2022, 10: 810327. doi: 10.3389/fcell.2022.810327
[10] CHEN Y F, LI X T, WANG S Y, et al. Targeting iron metabolism and ferroptosis as novel therapeutic approaches in cardiovascular diseases[J]. Nutrients, 2023, 15(3): 591. doi: 10.3390/nu15030591
[11] TIAN H, XIONG Y H, ZHANG Y, et al. Activation of NRF2/FPN1 pathway attenuates myocardial ischemia-reperfusion injury in diabetic rats by regulating iron homeostasis and ferroptosis[J]. Cell Stress Chaperones, 2021, 27(2): 149-164.
[12] WU H, LIU Q, SHAN X Y, et al. ATM orchestrates ferritinophagy and ferroptosis by phosphorylating NCOA4[J]. Autophagy, 2023, 19(7): 2062-2077. doi: 10.1080/15548627.2023.2170960
[13] FANG Y Y, CHEN X C, TAN Q Y, et al. Inhibiting ferroptosis through disrupting the NCOA4-FTH1 interaction: a new mechanism of action[J]. ACS Cent Sci, 2021, 7(6): 980-989. doi: 10.1021/acscentsci.0c01592
[14] LIANG D G, FENG Y, ZANDKARIMI F, et al. Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones[J]. Cell, 2023, 186(13): 2748-2764, e22. doi: 10.1016/j.cell.2023.05.003
[15] JIANG X J, STOCKWELL B R, CONRAD M. Ferroptosis: mechanisms, biology and role in disease[J]. Nat Rev Mol Cell Biol, 2021, 22(4): 266-282. doi: 10.1038/s41580-020-00324-8
[16] LIU J, KANG R, TANG D L. Signaling pathways and defense mechanisms of ferroptosis[J]. FEBS J, 2022, 289(22): 7038-7050. doi: 10.1111/febs.16059
[17] DOLL S, PRONETH B, TYURINA Y Y, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition[J]. Nat Chem Biol, 2017, 13(1): 91-98. doi: 10.1038/nchembio.2239
[18] TANG D L, KROEMER G. Peroxisome: the new player in ferroptosis[J]. Signal Transduct Target Ther, 2020, 5(1): 273. doi: 10.1038/s41392-020-00404-3
[19] 刘丹勇, 夏正远, 韩荣辉, 等. 心肌缺血再灌注损伤机制研究的回顾与展望[J]. 中国动脉硬化杂志, 2020, 28(12): 1013-1019. https://www.cnki.com.cn/Article/CJFDTOTAL-KDYZ202012002.htm [20] RODRIGO R, GONZÁLEZ-MONTERO J, SOTOMAYOR C G. Novel combined antioxidant strategy against hypertension, acute myocardial infarction and postoperative atrial fibrillation[J]. Biomedicines, 2021, 9(6): 620. doi: 10.3390/biomedicines9060620
[21] ZINDEL J, KUBES P. DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation[J]. Annu Rev Pathol, 2020, 15: 493-518. doi: 10.1146/annurev-pathmechdis-012419-032847
[22] WANG J, LIU Y, LIU Y, et al. Recent advances in nanomedicines for imaging and therapy of myocardial ischemia-reperfusion injury[J]. J Control Release, 2023, 353: 563-590. doi: 10.1016/j.jconrel.2022.11.057
[23] HEUSCH G. Myocardial ischaemia-reperfusion injury and cardioprotection in perspective[J]. Nat Rev Cardiol, 2020, 17(12): 773-789. doi: 10.1038/s41569-020-0403-y
[24] IBÁÑEZ B, HEUSCH G, OVIZE M, et al. Evolving therapies for myocardial ischemia/reperfusion injury[J]. J Am Coll Cardiol, 2015, 65(14): 1454-1471. doi: 10.1016/j.jacc.2015.02.032
[25] SPARVERO L J, TIAN H, AMOSCATO A A, et al. Direct mapping of phospholipid ferroptotic death signals in cells and tissues by gas cluster ion beam secondary ion mass spectrometry (GCIB-SIMS)[J]. Angew Chem Int Ed, 2021, 60(21): 11784-11788. doi: 10.1002/anie.202102001
[26] TANG L J, LUO X J, TU H, et al. Ferroptosis occurs in phase of reperfusion but not ischemia in rat heart following ischemia or ischemia/reperfusion[J]. Naunyn Schmiedebergs Arch Pharmacol, 2021, 394(2): 401-410. doi: 10.1007/s00210-020-01932-z
[27] YAN H F, ZOU T, TUO Q Z, et al. Ferroptosis: mechanisms and links with diseases[J]. Signal Transduct Target Ther, 2021, 6(1): 49. doi: 10.1038/s41392-020-00428-9
[28] LV Z Q, WANG F E, ZHANG X F, et al. Etomidate attenuates the ferroptosis in myocardial ischemia/reperfusion rat model via Nrf2/HO-1 pathway[J]. Shock, 2021, 56(3): 440-449. doi: 10.1097/SHK.0000000000001751
[29] KWON M Y, PARK E, LEE S J, et al. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death[J]. Oncotarget, 2015, 6(27): 24393-24403. doi: 10.18632/oncotarget.5162
[30] FAN Z Y, CAI L L, WANG S N, et al. Baicalin prevents myocardial ischemia/reperfusion injury through inhibiting ACSL4 mediated ferroptosis[J]. Front Pharmacol, 2021, 12: 628988. doi: 10.3389/fphar.2021.628988
[31] LI W Y, LI W, WANG Y, et al. Inhibition of DNMT-1 alleviates ferroptosis through NCOA4 mediated ferritinophagy during diabetes myocardial ischemia/reperfusion injury[J]. Cell Death Discov, 2021, 7(1): 267. doi: 10.1038/s41420-021-00656-0
[32] LEI D Y, LI B, ISA Z, et al. Hypoxia-elicited cardiac microvascular endothelial cell-derived exosomal miR-210-3p alleviate hypoxia/reoxygenation-induced myocardial cell injury through inhibiting transferrin receptor 1-mediated ferroptosis[J]. Tissue Cell, 2022, 79: 101956. doi: 10.1016/j.tice.2022.101956
[33] TANG L J, ZHOU Y J, XIONG X M, et al. Ubiquitin-specific protease 7 promotes ferroptosis via activation of the p53/TfR1 pathway in the rat hearts after ischemia/reperfusion[J]. Free Radic Biol Med, 2021, 162: 339-352. doi: 10.1016/j.freeradbiomed.2020.10.307
[34] LAKHAL-LITTLETON S, WOLNA M, CHUNG Y J, et al. An essential cell-autonomous role for hepcidin in cardiac iron homeostasis[J]. Elife, 2016, 5: e19804. doi: 10.7554/eLife.19804
[35] QIU M L, YAN W, LIU M M. YAP facilitates NEDD4L-mediated ubiquitination and degradation of ACSL4 to alleviate ferroptosis in myocardial ischemia-reperfusion injury[J]. Can J Cardiol, 2023, 39(11): 1712-1727. doi: 10.1016/j.cjca.2023.07.030
[36] CAI W B, LIU L, SHI X L, et al. Alox15/15-HpETE aggravates myocardial ischemia-reperfusion injury by promoting cardiomyocyte ferroptosis[J]. Circulation, 2023, 147(19): 1444-1460. doi: 10.1161/CIRCULATIONAHA.122.060257
[37] LEE Y S, LEE D H, CHOUDRY H A, et al. Ferroptosis-induced endoplasmic reticulum stress: cross-talk between ferroptosis and apoptosis[J]. Mol Cancer Res, 2018, 16(7): 1073-1076. doi: 10.1158/1541-7786.MCR-18-0055
[38] LI W Y, LI W, LENG Y, et al. Ferroptosis is involved in diabetes myocardial ischemia/reperfusion injury through endoplasmic reticulum stress[J]. DNA Cell Biol, 2020, 39(2): 210-225. doi: 10.1089/dna.2019.5097
[39] ZHOU Y Q, ZHOU H X, HUA L, et al. Verification of ferroptosis and pyroptosis and identification of PTGS2 as the hub gene in human coronary artery atherosclerosis[J]. Free Radic Biol Med, 2021, 171: 55-68. doi: 10.1016/j.freeradbiomed.2021.05.009
[40] ZHAO K, CHEN X S, BIAN Y J, et al. Broadening horizons: The role of ferroptosis in myocardial ischemia-reperfusion injury[J]. Naunyn Schmiedebergs Arch Pharmacol, 2023, 396(10): 2269-2286. doi: 10.1007/s00210-023-02506-5
[41] XU J F, ZHANG M H, LIU F, et al. Mesenchymal stem cells alleviate post-resuscitation cardiac and cerebral injuries by inhibiting cell pyroptosis and ferroptosis in a swine model of cardiac arrest[J]. Front Pharmacol, 2021, 12: 793829. doi: 10.3389/fphar.2021.793829
[42] YAN N, XU Z P, QU C H, et al. Dimethyl fumarate improves cognitive deficits in chronic cerebral hypoperfusion rats by alleviating inflammation, oxidative stress, and ferroptosis via NRF2/ARE/NF-κB signal pathway[J]. Int Immunopharmacol, 2021, 98: 107844. doi: 10.1016/j.intimp.2021.107844
-
期刊类型引用(1)
1. 郭英惠,代红燕,姚雪萍,任芳,尹娜,左晓婷. Klotho蛋白对大鼠心肌缺血-再灌注损伤的作用机制探究. 现代生物医学进展. 2024(19): 3616-3618+3644 .  百度学术
百度学术
其他类型引用(0)
 下载:
下载:
计量
- 文章访问数: 193
- HTML全文浏览量: 140
- PDF下载量: 29
- 被引次数: 1
 苏公网安备 32100302010246号
苏公网安备 32100302010246号