| Citation: |
HUANG Saisai, ZHAO Cheng, LIANG Jun, LU Xianyan, WANG Shiying. Mechanism of macrophage extracellular traps induced activation of fibroblast-like synoviocytes in the pathogenesis of rheumatoid arthritis[J]. Journal of Clinical Medicine in Practice, 2022, 26(23): 97-102. DOI: 10.7619/jcmp.20221368

|
To investigate the role of macrophage extracellular traps (METs) on fibroblast-like synoviocytes (FLSs) and related mechanisms.
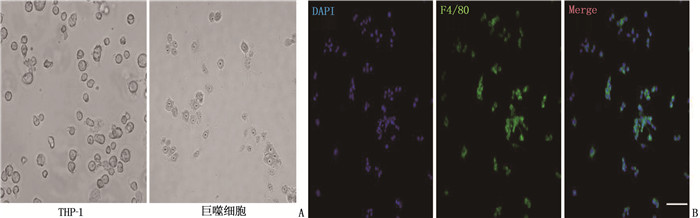
A human acute monocytic leukemia cell (THP-1) line was used to induce macrophages in vitro, and lipopolysaccharide (LPS) and interferon γ(IFN-γ) were used to induce METs; Sytox was used to mark the level of leaked DNA; Western blot and immunofluorescence were used to detect NE and MPO in METs. After purification of METs protein components, human FLSs cell lines were divided into control group, METs group, C29 group, IAXO-102 group and C29+IAXO-102 combined intervention group. Except for the control group, purified METs protein components were added to FLSs medium in the other groups. Toll-like receptor (TLR) 2 inhibitor C29 was added to the C29 group, TLR4 inhibitor IAXO-102 was added to the IAXO-102 group, and C29 and IAXO-102 were added to the C29+IAXO-102 combined intervention group. After 24 h, the viability, migration and invasion ability of FLSs cells were detected. The levels of interleukin-6 (IL-6), matrix metalloproteinase 1 (MMP-1), matrix metalloproteinase 3 (MMP-3) and matrix metalloproteinase 13 (MMP-13) in the culture supernatant were determined by enzyme-linked immunosorbent assay (ELISA).
LPS induced METs ability was significantly stronger than IFN-γ (P < 0.05). METs protein significantly enhanced FLSs cell viability, synovial cell migration and invasion ability (P < 0.05); compared with the control group, IL-6, MMP-1, MMP-3 and MMP-13 in the supernatant of METs protein treatment group were significantly increased (P < 0.05). C29 or IAXO-102 alone or their combination could significantly reduce the viability, migration and invasion ability of synovial cells (P < 0.05); compared with METs group, the levels of IL-6, MMP-1, MMP-3 and MMP-13 in the combined intervention group were significantly decreased (P < 0.05), and the inhibitory effect of IAXO-102 was significantly stronger than that of C29 (P < 0.05).
METs proteins as damage-related model molecules are mainly involved in RA pathogenesis by activating TLR4 to further activate FLSs.
| [1] |
WASSERMAN A M. Diagnosis and management of rheumatoid arthritis[J]. Am Fam Physician, 2011, 84(11): 1245-1252.
|
| [2] |
SCHERER H U, HÄUPL T, BURMESTER G R. The etiology of rheumatoid arthritis[J]. J Autoimmun, 2020, 110: 102400. doi: 10.1016/j.jaut.2019.102400
|
| [3] |
BERGOT A S, GIRI R, THOMAS R. The microbiome and rheumatoid arthritis[J]. Best Pract Res Clin Rheumatol, 2019, 33(6): 101497. doi: 10.1016/j.berh.2020.101497
|
| [4] |
PAPAYANNOPOULOS V. Neutrophil extracellular traps in immunity and disease[J]. Nat Rev Immunol, 2018, 18(2): 134-147. doi: 10.1038/nri.2017.105
|
| [5] |
DOSTER R S, ROGERS L M, GADDY J A, et al. Macrophage extracellular traps: a scoping review[J]. J Innate Immun, 2018, 10(1): 3-13. doi: 10.1159/000480373
|
| [6] |
BRINKMANN V, REICHARD U, GOOSMANN C, et al. Neutrophil extracellular traps kill bacteria[J]. Science, 2004, 303(5663): 1532-1535. doi: 10.1126/science.1092385
|
| [7] |
MÖLLERHERM H, VON KÖCKRITZ-BLICKWEDE M, BRANITZKI-HEINEMANN K. Antimicrobial activity of mast cells: role and relevance of extracellular DNA traps[J]. Front Immunol, 2016, 7: 265.
|
| [8] |
JE S, QUAN H L, YOON Y, et al. Mycobacterium massiliense induces macrophage extracellular traps with facilitating bacterial growth[J]. PLoS One, 2016, 11(5): e0155685. doi: 10.1371/journal.pone.0155685
|
| [9] |
NYGAARD G, FIRESTEIN G S. Restoring synovial homeostasis in rheumatoid arthritis by targeting fibroblast-like synoviocytes[J]. Nat Rev Rheumatol, 2020, 16(6): 316-333. doi: 10.1038/s41584-020-0413-5
|
| [10] |
MCINNES I B, SCHETT G. The pathogenesis of rheumatoid arthritis[J]. N Engl J Med, 2011, 365(23): 2205-2219. doi: 10.1056/NEJMra1004965
|
| [11] |
MICHELS K R, LUKACS N W, FONSECA W. TLR activation and allergic disease: early life microbiome and treatment[J]. Curr Allergy Asthma Rep, 2018, 18(11): 61. doi: 10.1007/s11882-018-0815-5
|
| [12] |
KYBURZ D, RETHAGE J, SEIBL R, et al. Bacterial peptidoglycans but not CpG oligodeoxynucleotides activate synovial fibroblasts by toll-like receptor signaling[J]. Arthritis Rheum, 2003, 48(3): 642-650. doi: 10.1002/art.10848
|
| [13] |
ALSOUSI A A, IGWE O J. Redox-active trace metal-induced release of high mobility group box 1(HMGB1) and inflammatory cytokines in fibroblast-like synovial cells is Toll-like receptor 4 (TLR4) dependent[J]. Biochim Biophys Acta Mol Basis Dis, 2018, 1864(11): 3847-3858. doi: 10.1016/j.bbadis.2018.08.029
|
| [14] |
WU R, LONG L, CHEN Q Q, et al. Effects of Tim-3 silencing on the viability of fibroblast-like synoviocytes and lipopolysaccharide-induced inflammatory reactions[J]. Exp Ther Med, 2017, 14(3): 2721-2727. doi: 10.3892/etm.2017.4819
|
| 1. |
郑执一,曹鹏飞,陈文奇,王锐,乔建龙,邵花香,李华玲. ANGPTL4的作用及机制. 生命的化学. 2024(05): 890-899 .

|
© 2020 《实用临床医药杂志》编辑部
Address: 江苏省扬州市江阳中路136号,扬州大学江阳路北校区14号楼201室China Pos: 225009Tel: 0514-87978917、87978989、87978807
Supported by:
Beijing Renhe Information Technology Co., Ltd.

 苏公网安备 32100302010246号
苏公网安备 32100302010246号 DownLoad:
DownLoad: